GW始まってしまいましたね。
ブログの更新をだいぶ怠っていたので、ちゃっかり更新させて頂きます。
今日はPythonでscRNA-seq解析。Python実装のscRNA解析ツールといえばScanpyがまず思いつきます。
Seuratに比べてそこまで使われていない印象ですが、機能的には十分すぎる上にチュートリアルも充実してるのでPythonユーザーにはこちらの方が扱いやすいかもしれません。
Let's scanpy!
Scanpyとは
Scanpyはsingle-cell RNAの発現量データを解析するためのツールです。 データの前処理、可視化、クラスタリング、疑似系譜解析、発現変動解析なんかが可能です。
Seuratを踏襲しているのか、ところどころでSeuratぽさが散見されます。
論文はGenome Biology誌に2018/2/6にpublishされています。
2019/2/5にversion 1.4.0がリリースされました。
今回はこのScanpy version 1.4.0を使用して一連の解析を見ていきたいと思います。
チュートリアル
Clustering 3K PBMCに沿って、解析のチュートリアルを行っていきます。
このチュートリアルは、SeuratのチュートリアルをScanpyを使ってやってみた的なものになっています。
ver3に対応済みのようなので(参照)
ver 3に直しながらチュートリアルを行っていきます。
解析の前準備
まずは解析を行うディレクトリを作成し、移動しましょう。
$ mkdir scanpy_tutrial $ cd scanpy_tutrial
データのダウンロードはwgetコマンドで行います。
詳細は以下の通り、HiSeq4000でシーケンスした健常者のPBMCです。
・713 cells detected
・Sequenced on Illumina HiSeq4000 with approximately 63,000 reads per cell
・28bp read1 (16bp Chromium barcode and 12bp UMI), 91bp read2 (transcript), and 8bp I7 sample barcode
・run with --expect-cells=1000
・This dataset was generated using a pre-release set of TotalSeq-B antibodies. The barcode sequences have since changed. Please refer to https://www.biolegend.com/totalseq for the latest conjugated barcode information.
引用:https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_1k_protein_v3
リンクが切れていた場合は10Xのサイトから適当なversion 3のデータをダウンロードしてください。
$ wget http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_protein_v3/pbmc_1k_protein_v3_filtered_feature_bc_matrix.tar.gz
インストール
scanpyのインストールを行います。
condaでもpipでも入るのでお好きなものをご利用ください。
condaを使用
conda install conda-forge scanpy python-igraph leidenalg
pipを使用
PyPIで公開されているため、pip コマンドによりインストール可能です。
pip install scanpy
pipenvを使用
書くまでもないですが以下の通りです。
pipenv install scanpy
チュートリアルを進める際は、その他にnumpy及びpandasを使用するので同様の方法でインストールしてください。
データの読み込み
ログレベルを1 ~ 3から選択、出力ファイルを指定します。
import numpy as np import pandas as pd import scanpy as sc sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3) sc.logging.print_versions() results_file = './write/pbmc3k.h5ad' # the file that will store the analysis results
read_10x_mtxメソッドは
最近のバージョンアップ にてcellranger v3出力データの読み込みに対応しています。
引数には.mtx を含んだディレクトリを指定しましょう。
>>> adata = sc.read_10x_mtx(
'./data/filtered_feature_bc_matrix/',
var_names='gene_symbols',
cache=True)
データを読み込んだオブジェクトはAnnDataというクラスになっています。
scanpyでは基本的にこのオブジェクトを操作して様々な解析を行います。
>>> anndata.base.AnnData
まず格納された細胞数、遺伝子数を確認しておきましょう。
各時点において使用することのできるデータは、オブジェクトの__str__で確認できるように定義されています。
つまりは単純にオブジェクトの変数名を入力すればよいです。
>>> adata
View of AnnData object with n_obs × n_vars = 713 × 33538
var: 'gene_ids', 'feature_types'
713 × 33538
713 細胞 × 33538遺伝子を意味しています。
先ほど引用した10xのページ上の細胞数と合致していますね。
・713 cells detected
・Sequenced on Illumina HiSeq4000 with approximately 63,000 reads per cell
・28bp read1 (16bp Chromium barcode and 12bp UMI), 91bp read2 (transcript), and 8bp I7 sample barcode
・run with --expect-cells=1000
・This dataset was generated using a pre-release set of TotalSeq-B antibodies. The barcode sequences have since changed. Please refer to https://www.biolegend.com/totalseq for the latest conjugated barcode information.
引用:https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_1k_protein_v3
QC
データを読み込んだらまずはQuality Check 通称QC です。
本来は様々な観点からデータを眺める必要があるのですが、ここではチュートリアルに沿ってBar Plotを描画するのみにとどめます。
フィルタリング後に再度QCを行います。
Bar Plotの描画
以下のコマンドでは、高発現量の遺伝子上位20を描画します。
>>> sc.pl.highest_expr_genes(adata, n_top=20)

MALAT1が高発現していますね。
この遺伝子は転移性肺癌で高発現しているとか何とか。
後はMT-から始まるミトコンドリアRNAがいくつか発現しています。
Prefiltering
明らかに発現細胞数が少ない遺伝子や発現遺伝子数が少ない細胞は前もって除いておきます。
今回は発現遺伝子数が200細胞未満の細胞、発現している細胞数が3未満の遺伝子を除去します。
>>> sc.pp.filter_cells(adata, min_genes=200) >>> sc.pp.filter_genes(adata, min_cells=3) filtered out 15 cells that have less than 200 genes expressed filtered out 19272 genes that are detected in less than 3 cells
adata内のデータが置き換えられています。
出力の通り、15細胞、19272遺伝子がフィルターアウトされています。
確認すると、
>>> adata
AnnData object with n_obs × n_vars = 698 × 14266
obs: 'n_genes'
var: 'gene_ids', 'feature_types', 'n_cells'
698 × 14266
確かに15細胞、19272遺伝子がフィルターアウトされていることが確認できます。
アノテーション
Prefilteringを行った後、さらにデータに合わせた条件にてフィルタリングを行います。
その前にデータを表す有用な特徴量を算出し、アノテーションしておきましょう。
ミトコンドリアRNA割合の算出
ミトコンドリアRNAの割合は細胞のクオリティーを判定する上で重要な指標となります。
High numbers of mitochondrial transcripts are indicators of cell stress,41 and therefore cells with elevated mitochondrial gene expression are often not included in the analysis, because most experiments will not benefit from clustering cells based on stress levels. However, just as with number of transcripts, this parameter is highly dependent on the tissue type and the questions being investigated. For example, 30% of total mRNA in the heart is mitochondrial due to high energy needs of cardiomyocytes, compared with 5% or less in tissues with low energy demands.42 For instance, 30% mitochondrial mRNA is representative of a healthy heart muscle cell, but would represent a stressed lymphocyte.
An Introduction to the Analysis of Single-Cell RNA-Sequencing Data
Aisha A. AlJanahi,1,2 Mark Danielsen,2 and Cynthia E. Dunbar, Translational Stem Cell Biology Branch, Published:July 15, 201830066-4)
高ければ高いほど死細胞や欠陥のある細胞の割合が高いことを示唆しているためです。
細胞中のミトコンドリアRNAの割合及び発現UMIカウント数を以下のように追加します。
単純に「MT-」から始まる遺伝子をミトコンドリアRNAとし、各細胞ごとにミトコンドリアRNAの発現量を全遺伝子の総発現量で割っています。
ここではnumpyのA1メソッドを使用することでMatrixを平滑化しています。
# 「MT-」から始まる遺伝子を >>> mito_genes = adata.var_names.str.startswith('MT-') # 各細胞ごとにミトコンドリア関連遺伝子の発現量を全遺伝子の総発現量で割る >>> adata.obs['percent_mito'] = np.sum(adata[:, mito_genes].X, axis=1).A1 / np.sum(adata.X, axis=1).A1 # adataオブジェクトの`n_counts`属性に総UMI数のアノテーションを格納 >>> adata.obs['n_counts'] = adata.X.sum(axis=1).A1
Violin Plot の描画
フィルタリングの閾値を決定するために、再度QCを行います。
ここでは細胞の密度分布を可視化する際に有用な Violin Plot を描画します。
>>> sc.pl.violin(adata, ['n_genes', 'n_counts', 'percent_mito'], jitter=0.4, multi_panel=True)

Scatter Plot の描画
細胞のクオリティーを判別する際にはViolin PlotのほかにScatter Plotも有用です。
Scatter Plotでは二次元空間でプロットを行うことができるため、以下のような考察を行うことができます。
- UMIカウントに対し過剰にミトコンドリアRNAを発現しているような細胞を死細胞とみなす
- 総遺伝子数がUMIカウントに対し過剰に高くなっているようなデータはdoubletとみなす

フィルタリング
上記で可視化したプロットを参考に、さらにフィルタリングを行います。 Violin plot の結果から、今回は
- 発現遺伝子数 < 5000
- 発現ミトコンドリア関連遺伝子割合 < 0.25
をフィルタリングの条件とします。
>>> adata = adata[adata.obs['n_genes'] < 5000, :] >>> adata = adata[adata.obs['percent_mito'] < 0.25, :]
細胞数を確認すると、
細胞数は698 → 570に細胞数が減っていることがわかります。
>>> adata View of AnnData object with n_obs × n_vars = 570 × 14266 obs: 'n_genes', 'percent_mito', 'n_counts' var: 'gene_ids', 'feature_types', 'n_cells'
正規化
細胞間で値の比較を行えるように、データの正規化を行います。
normalize_per_cellメソッドを使用すれば、指定したリード数当たりの発現量に変換することができます。
>>> sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
さらにlog1pメソッドにより対数変換を行うことが可能です。
>>> sc.pp.log1p(adata)
HVG (Highly Variable Genes)の検出
highly_variable_genesメソッドを使用すると、平均発現量及び分散から有意な遺伝子を同定することができます。
>>> sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
可視化も簡単に行うことができます。
>>> sc.pl.highly_variable_genes(adata)

ここでマーキングした有意遺伝子の情報はBoolean型で格納されているため、
以下のコマンドで有意遺伝子のみにフィルタリングすることができます。
>>> adata = adata[:, adata.var['highly_variable']] View of AnnData object with n_obs × n_vars = 570 × 2806 obs: 'n_genes', 'percent_mito', 'n_counts' var: 'gene_ids', 'feature_types', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
発現遺伝子数のみに着目するため、n_counts及びpercent_mitoの影響を除いた値について標準偏差10以下の細胞のみにフィルタリングを行います。
>>> sc.pp.regress_out(adata, ['n_counts', 'percent_mito']) >>> sc.pp.scale(adata, max_value=10)
次元圧縮(PCA)
次元圧縮を行うことで、高次元のデータを低次元で可視化することが可能となります。
PCAは次元圧縮手法の中でも最もメジャーな手法です。
第一主成分の分散を最大化し、続く主成分はそれまでに決定した主成分と直交するような軸を分散が最大となるように選ぶことで、データの次元を落とすことができます。
PCAを行い、CST3遺伝子の発現量についてPC1、PC2の2軸で可視化を行います。
>>> sc.tl.pca(adata, svd_solver='arpack') >>> sc.pl.pca(adata, color='CST3')

有意な軸数は俗にいうElbow Plot)により判別することができます。
pca_variance_ratioメソッドを使用することで、Elbow Plotを描画することができます。
>>> sc.pl.pca_variance_ratio(adata, log=True)

ここで、writeメソッドにより中間データを保存します。
>>> adata.write(results_file)
PCAの結果から近傍グラフの算出を行います。 Seuratと同じアルゴリズムのようですね。 チュートリアルに則り、主成分40次元に対しn=10で実行します。
>>> sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
次元圧縮 (UMAP)
scanpyでは、Seurat でも実装されているUMAP (Uniform Manifold Approximation and Projection) を使用することができます。
The algorithm implementing this technique is demonstrably faster than t-SNE and provides better scaling. This allows us to generate high quality embeddings of larger data sets than had previously been attainable. Thee use and effectiveness of UMAP in various scientific fields demonstrates the strength of the algorithm.
UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction, Leland McInnes, John Healy, James Melville, Submitted on 9 Feb 2018
t-SNEよりも早く良い性能だとか。
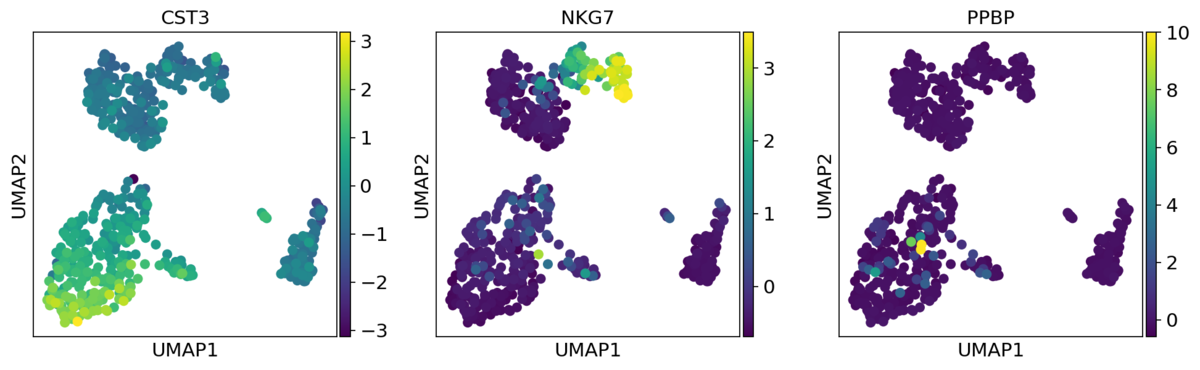
CST3、NKG7、PPBPの3遺伝子について
>>> sc.tl.umap(adata) >>> sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])

raw属性が格納されているとこちらのデータが優先して使用されます。
use_rawオプションを指定することでスケーリング後のデータを使用することもできます。
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
クラスタリング
Seuratを含め、scRNA解析ツールでは、Louvain Modularity を応用したクラスタリングが採用されることが多いです。
この手法はコミュニティー検出とそれに続くモジュール最適化で構成されています。
近傍グラフは先ほど算出したため、これを使用してモジュール最適化を行います。
以下では igraph(0.7.1)を使用します。
$ pipenv install python-igraph louvain
ここでは内部的にlouvainパッケージを使用しています。
>>> sc.tl.louvain(adata)
louvainクラスタリングの結果及びCST3、NKG7遺伝子の発現量をUMAP上にプロットします。
>>> sc.pl.umap(adata, color=['louvain', 'CST3', 'NKG7'])

再度結果を保存しておきましょう。
>>> adata.write(results_file)
t検定により各クラスターにおいて有意な遺伝子をプロットします。
プロットする遺伝子数はn_genesで指定します。今回は25遺伝子についてプロットを行います。
Wilcoxonの順位和検定も使用可能です。
diffxpyを使用すれば、その他著名な手法であるMAST、limma、DESeq2についても利用可能です。
# T-test sc.tl.rank_genes_groups(adata, 'louvain', method='t-test') sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

# Wilcoxon sc.tl.rank_genes_groups(adata, 'louvain', method='wilcoxon') sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

上述の解析はいずれも単変量解析となりますが、多変量解析も用いることができます。
ロジスティック回帰を使用する際はmethod='logreg'を指定します。
# logistic regression sc.tl.rank_genes_groups(adata, 'louvain', method='logreg') sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

セルタイプの特定
各クラスターにおいて有意に発現している遺伝子から、クラスターのセルタイプを特定します。
今回の場合、下記のような結果になります。重複するセルタイプが出現してしまっているのはクラスタリングがうまくいっていないせいです。
本来であればパラメータを調整し、セルタイプごとにクラスターが形成されるようにクラスタリングを行う必要があります。
| Louvain Group | Markers | Cell Type |
|---|---|---|
| 0 | CD14, LYZ | CD14+ Monocytes |
| 1 | IL7R CD4 | T cells |
| 2 | MS4A1 | B cells |
| 3 | TCF7 | T cells |
| 4 | CST3 | Dendritic Cells |
| 5 | CST3 | Dendritic Cells |
| 6 | CD8A | CD8 T cells |
| 7 | GNLY, NKG7 | NK cells |
| 8 | LST1, MS4A7 | Leukocyte, Monocytes |
>>> pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | S100A8 | S100A4 | IGKC | CD7 | LGALS2 | CDKN1A | CCL5 | GNLY | FCGR3A |
| 2 | S100A12 | IL7R | CD74 | LEF1 | HLA-DPB1 | ZFP36L1 | GZMK | NKG7 | CDKN1C |
| 3 | S100A9 | ITGB1 | HLA-DRA | JUNB | YBX3 | MYADM | NKG7 | KLRD1 | MS4A7 |
| 4 | CXCL8 | IL32 | IGHM | GYPC | CPVL | ATF3 | GZMA | KLRF1 | LST1 |
| 5 | GCA | LTB | HLA-DRB1 | SLC2A3 | HLA-DQA2 | POU2F2 | KLRB1 | FCER1G | SMIM25 |
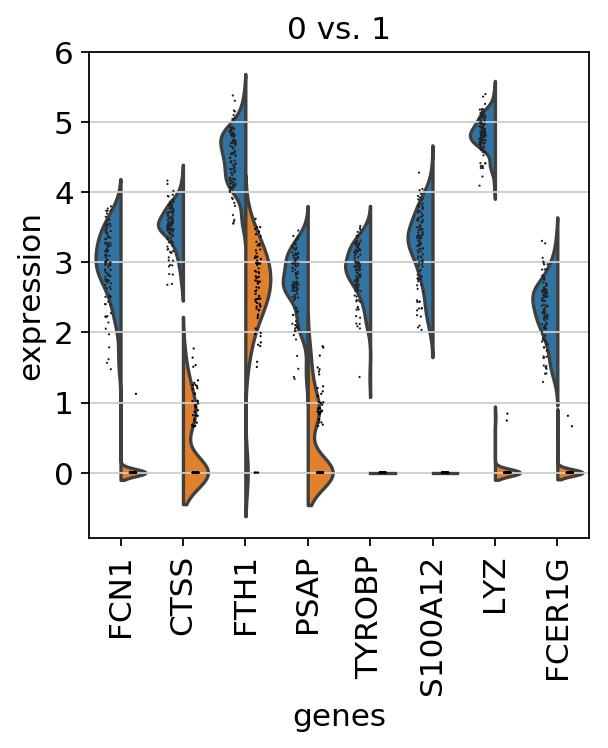
2クラスター間の比較
クラスター0とクラスター1を比較する。
ここでもmethodを指定して検定手法を選択することができます。
>>> sc.tl.rank_genes_groups(adata, 'louvain', groups=['0'], reference='1', method='wilcoxon') >>> sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)

Violin Plot
発現量の比較を含めてプロットする際はrank_genes_groups_violinメソッドを使用します。
>>> sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
クラスターごとの遺伝子発現量をバイオリンプロットで描画する際は以下のように行います。
>>> sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='louvain')

以上!
こんな感じです。あくまでチュートリアルをなぞっただけなのでまだまだ機能は豊富なscanpyとなっています。
PythonでscRNA-seq解析を行いたい方はぜひお試しあれ。